01 Jul 2019 Mutaciones genómicas germinales y susceptibilidad al desarrollo de leucemias
Julia Suárez-González1,2, Cristina Andrés-Zayas1,2, Gabriela Rodríguez-Macías3, María Chicano2,3, Diego Carbonell2,3, Paula Muñiz2,3, Nieves Dorado2,3, Pascual Balsalobre2,3, Mi Kown2,3, José Luis Díez-Martín2,3, Ismael Buño1,2,3*, Carolina Martínez-Laperche2,3*.
(*) Último autor, contribución idéntica.
1Unidad de Genómica, Hospital G.U. Gregorio Marañón/Instituto de Investigación
Sanitaria Gregorio Marañón (IiSGM).
2Instituto de Investigación Sanitaria Gregorio Marañón (IiSGM).
3Servicio de Hematología, Hospital G.U. Gregorio Marañón.
Título corto: Susceptibilidad genómica al desarrollo de leucemias
Palabras clave: cáncer, leucemia, neoplasia mieloide, predisposición genética, mutación germinal, genómica.
*Autor para la Correspondencia: Carolina Martínez-Laperche e Ismael Buño. Laboratorio de Genética Hematológica Servicio de Hematología Hospital G.U. Gregorio Marañón Instituto de Investigación Sanitaria Gregorio Marañón (IiSGM) C/ Doctor Esquerdo 46 28009 Madrid Correo electrónico: cmlaperchehgugm@gmail.com ismaelbuno@gmail.com
Introducción
En los últimos años, la genética ha revolucionado nuestro conocimiento sobre el cáncer. Es conocido que los cánceres son enfermedades genéticas de las células somáticas, si bien una parte de ellos está muy condicionada por mutaciones genéticas heredadas en la línea germinal que se comportan como rasgos mendelianos.1 Durante mucho tiempo las leucemias mieloides agudas (LMA) y los síndromes mielodisplásicos (SMD) han sido considerados eventos de novo, que ocurrían de manera aleatoria en la población general. Únicamente, los síndromes relacionados con fallo de médula ósea, enfermedades teloméricas y síndrome de Down se asociaban con predisposición al desarrollo de SMD y/o LMA congénita. Sin embargo, en los últimos años, se han definido un subtipo de mutaciones constitucionales que le otorgan al individuo una susceptibilidad al desarrollo de neoplasias hematológicas mieloides.2 Este grupo de síndromes de predisposición al desarrollo de neoplasias mieloides forman parte de una nueva entidad en la última revisión de la clasificación de las neoplasias hematológicas realizada por la Organización Mundial de la Salud (World Health Organization; WHO, 2016; Tabla 1).3
Tabla 1. Clasificación de los síndromes de predisposición al desarrollo de neoplasias mieloides (OMS 2016).3
Neoplasia mieloide con mutación en línea germinal sin enfermedad o disfunción orgánica pre-existente.
LMA con mutación germinal de CEBP
Neoplasia mieloide con mutación germinal de DDX41
Neoplasia mieloide con mutación en línea germinal y desórdenes plaquetarios preexistentes
Neoplasia mieloide con mutación germinal en RUNX1
Neoplasia mieloide con mutación germinal en ANKRD26
Neoplasia mieloide con mutación germinal en ETV6
Neoplasias mieloides con mutación en línea germinal y otra disfunción orgánica
Neoplasia mieloide con mutación germinal en GATA2
Neoplasia mieloide asociada con síndromes de fallo de médula ósea
Neoplasia mieloide asociada con desórdenes de telómeros Leucemia mielomonocítica juvenil asociada con Neurofibromatosis, S. Noonan o Noonan-like
Neoplasia mieloide asociada con síndrome de Down
El reconocimiento de individuos con una susceptibilidad al cáncer heredada se basa, habitualmente, en la recogida cuidadosa de los antecedentes familiares para documentar la presencia o la ausencia de otros miembros de la familia con cánceres similares o relacionados. Además, hay algunas características que pueden sugerir un síndrome de susceptibilidad heredada al cáncer en una familia, tales como:
– Varios familiares próximos, de primero o segundo grado, con un cáncer frecuente o con cánceres relacionados.
– Dos miembros de la familia con el mismo cáncer raro.
– Edad de presentación inusualmente precoz.
– Tumores bilaterales en órganos pares.
– Tumores sincrónicos o sucesivos.
– Tumores en dos sistemas orgánicos diferentes en un individuo.
Neoplasias mieloides de origen germinal
Las neoplasias mieloides de origen germinal son trastornos autosómicos dominantes, cuya forma de aparición es muy heterogénea. 4,5 Algunos pacientes debutan con leucemia aguda mientras que otros, tienen alguna patología o disfunción orgánica preexistente. 4,5 Por otro lado, hay pacientes que tienen una clara agregación familiar de neoplasias hematológicas u otros tumores sólidos, mientras que otros no presentan antecedentes familiares, bien por la expresividad variable de la enfermedad o porque son eventos de novo. Esto hace que sea difícil la detección de los mismos. Es importante diferenciar aquellas neoplasias mieloides que tienen origen germinal de aquellas que aparecen espontáneamente o debido a la exposición a factores ambientales o químicos nocivos, ya que, es decisivo para el tratamiento y el seguimiento a largo plazo de los pacientes afectos. Incluso si el paciente no ha desarrollado una neoplasia, la presencia de disfunción orgánica y/o anomalías en el número y función de las plaquetas requiere un tratamiento específico.3 Además, aquellos pacientes que son considerados para someterse a un trasplante alogénico de progenitores hematopoyéticos, es muy importante la selección cuidadosa de los donantes en estas familias para evitar la re-introducción de una mutación deletérea en el paciente, ya que el empleo inadvertido de un donante afecto puede ocasionar un fallo del injerto o una leucemia en células del donante, definida como el desarrollo de una neoplasia de novo en las células del donante.6,7
Estudio genético
Son numerosos los genes asociados actualmente, con la predisposición al desarrollo de neoplasias mieloides (Figura 1). Cuando se sospecha que un individuo, diagnosticado de alguna neoplasia mieloide en la edad adulta, tiene una susceptibilidad heredada al desarrollo de neoplasia mieloides es necesario realizar un estudio que debe englobar, al menos, los genes CEBP , TP53, ANKRD26, ETV6, RUNX1, DDX41 y GATA2.
Figura 1. Genes asociados con predisposición al desarrollo de neoplasias mieloides
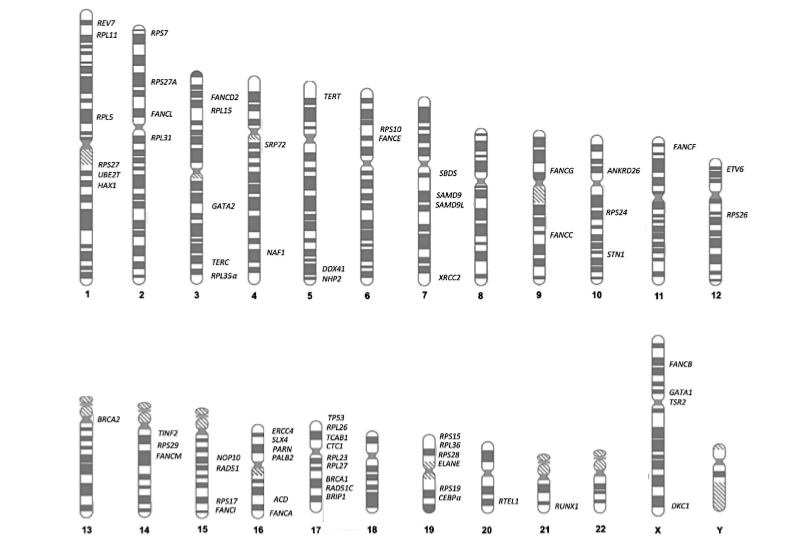
Los estudios pueden realizarse bien mediante secuenciación convencional Sanger o mediante secuenciación masiva de un panel de genes para la detección de mutaciones puntuales y de pequeños indels. Adicionalmente, para el estudio de deleciones o
duplicaciones del gen es necesario el empleo de técnicas específicas como el MLPA o la secuenciación masiva ya que estas alteraciones pueden pasar desapercibidas con las técnicas convencionales.
Cuando se realizan estas pruebas en pacientes con LMA o SMD es importante recordar que la sangre es un tejido que está dañado y que muchos de los genes mutados en línea germinal también pueden estar mutados somáticamente,8 lo que podría llevar a una mala interpretación de los resultados si las pruebas se realizan en la médula ósea o sangre periférica de los pacientes. Por esta razón, se recomienda el empleo de
fibroblastos obtenidos mediante una biopsia cutánea para la realización de estas pruebas. Los fibroblastos crecen en un cultivo in vitro y suelen tardar en crecer entre unas 4-6 semanas. Este es un tiempo prolongado, que puede no ser de utilidad en aquellos casos en los que se necesita tener un resultado para la toma de decisiones clínicas. En estas situaciones, se pueden obtener muestras de mucosa bucal o de
saliva. Sin embargo, hay que interpretar los resultados con cautela ya que ambas muestras pueden contener linfocitos contaminantes. Es posible, a su vez, estudiar en los potenciales donantes la variante sospechosa.
Consejo Genético
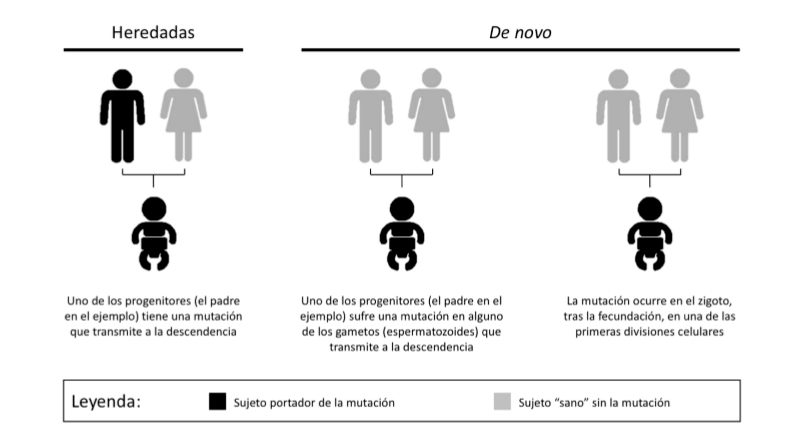
Es importante acudir a una consulta especializada antes y después de la realización dela prueba genética. Durante la consulta previa a la prueba genética, los pacientes y susfamiliares deben conocer las bases principales de la genética humana (Figura 2) y losdetalles que se conocen específicos del síndrome que se sospecha. Esta información esla base del proceso de consentimiento informado para la realización de pruebas genéticas, asegurándose que el paciente entiende los riesgos y los beneficios de laprueba, los posibles resultados de la prueba y el impacto de los mismos sobre sí mismo y sobre sus familiares.
Idealmente, es necesario realizar otra consulta de consejo genético tanto si los resultados son negativos o positivos. El consejo genético posterior a la prueba debe englobar la explicación sobre el significado y la implicación de los resultados sobre la
salud y recomendaciones basadas en sus resultados y en su historia familiar. En el caso de un resultado positivo en un paciente, también es importante discutir las implicaciones de esos resultados sobre miembros de la familia que pueden tener unriesgo de tener la misma alteración e informar a estos pacientes sobre su deber de compartir esta información con los miembros de la familia de riesgo. El consejo genético individual de estos miembros de la familia también es recomendable.
Otras implicaciones sobre la salud de los pacientes, los riesgos que tienen de desarrollar otras alteraciones no hematológicas, así como implicaciones en la sensibilidad al tratamiento, la posibilidad de protocolos de tratamiento alternativos en estos pacientes, el riesgo de recurrencia y la posibilidad de realizar un test genético preimplantacional también se deben abordar en el consejo genético.9
Figura 2. Mutaciones genéticas de origen germinal
Seguimiento
La prevención o la detección precoz de la neoplasia hematológica es el objetivo final de estos estudios, por lo que es recomendable, que todos los portadores de mutaciones se realicen un hemograma con recuento diferencial cada 6 meses. Si se observa alguna anomalía en el recuento, se repetirá 1-2 semanas más tarde y si el cambio persiste, será necesario realizar un aspirado de médula ósea.10
En todos los portadores de mutaciones debería evitarse la exposición a fármacos con toxicidad en médula ósea y hábitos tóxicos como el tabaco y el abuso de alcohol.7
Por otro lado, en aquellas familias que se observa una historia clínica consistente con la existencia de un síndrome familiar de desarrollo de SMD/LMA, pero que no se ha detectado ninguna mutación en los genes que definen los síndromes familiares conocidos, deberán tener un plan se seguimiento igual que el de aquellos en los que se le ha detectado la mutación y deberán considerarse los riesgo y beneficios de emplear un donante no emparentado en estos casos.11
Conclusiones
– Es importante que los clínicos a cargo de pacientes con SMD/LMA estén familiarizados con la existencia de estos síndromes que se asocian con desarrollo de neoplasias hematológicas en ciertas familias.
– La mayoría de los síndromes de predisposición al desarrollo de neoplasias mieloides son extraordinariamente raros en la población; pueden, no obstante, llegar a explicar en conjunto aproximadamente el 5-10% de todas las neoplasias hematológicas.
– El reconocimiento de estos síndromes es crucial para el manejo clínico de los pacientes con una susceptibilidad heredada y para sus familiares que pueden ser portadores.
– La incorporación de las nuevas tecnologías de secuenciación masiva a la práctica clínica está cambiando la aproximación al diagnóstico de numerosas patologías.
– El espectro de mutaciones en los genes asociados con predisposición al desarrollo de neoplasias mieloides es muy amplio, y las características moleculares de las diferentes mutaciones parecen influir en el riesgo variable de desarrollar SMD o LMA.
– Debemos conocer en detalle la arquitectura genética subyacente, lo que implica no sólo identificar los loci implicados, sino también las posibles interacciones entre ellos.
– En los próximos años, probablemente se vaya identificando un número mayor de genes implicados en estos síndromes de predisposición al desarrollo de neoplasias tanto mieloides como de otras neoplasias hematológicas y se irán definiendo los ya conocidos.
Referencias
1. Owen C, Barnett M, Fitzgibbon J. Familial myelodysplasia and acute myeloid leukaemia – a review. Br J Haematol 2008. 140(2):123.
2. Nickels EM, Soodalter J, Churpek JE, Godley LA. Recognizing familial myeloid leukemia in adults. Ther Adv Hematol 2013. 4(4):254.
3. Arber DA, Orazi A, Hasserjian R, thiele J, Borowitz MJ, Le Beau MM et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016. 127(20):2391.
4. Hahn C, Chong C, Carmichael C, Wilkins E, Brautigan P, Li X, et al. Heritable GATA2 mutations associated with familial myelodysplastic síndrome and acute myeloid leukemia. Nat Genet 2011. 43(10):1012.
5. Tawana K, Wang J, Renneville A, Bödör C, Hills R, Loveday C, et al. Disease evolution and outcomes in familial AML with germline CEBP mutations. Blood 2015. 126(10):1214.
6. Suárez-González J, Martínez-Laperche C, Kwon M, Balsalobre P, Carbonell D, Chicano M, et al. Donor Cell-Derived Hematologic Neoplasms after Hematopoietic Stem Cell Transplantation: A Systematic Review. Biol Blood Marrow Transplant. 2018 (en prensa). doi: 10.1016/j.bbmt.2018.01.033.
7. Suárez-González J, Martínez-Laperche C, Martínez N, Rodríguez-Macías G, Kwon M, Balsalobre P, et al. Whole exome sequencing reveals acquisition of mutations leading to the onset of donor cell leukemia after hematopoietic transplantation: a model of leukemogenesis. Leukemia 2018 (en prensa). DOI: 10.1038/s41375-018-0042-z
7. Churpek JE, Pyrtel K, Kanchi K-L, Shao J, Koboldt D, Miller CA, et al. Genomic analyses of germline and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood 2015. 126:2484.
8. Churpek JE, Lorenz R, Nedumgottil S, et al. Proposal for the clinical detection and management of patients and their family members with familial myelodysplastic síndrome/acute leukemia predisposition síndrome. Leuk Lymphoma 2013. 54:28-35.
9. Godley LA. Inherited predisposition to acute myeloid leukemia. Semin Hematol 2014. 51(4):306.
10. The University of Chicago Hematopoietic Malignancies Cancer Risk Team. How I diagnose and manage individuals at risk for inherited myeloid malignancies. Blood 2016. 128(14):1800-1813.