01 Jul 2019 La enfermedad de Alzheimer. El difícil equilibrio entre lo específico y lo genérico, lo posible y lo imposible
Adolfo Toledano
Instituto de Neurobiología Ramón y Cajal, CSIC
Introducción
Cuando existen más de un centenar de teorías sobre una enfermedad, basadas en más de cien mil importantes trabajos de investigación en los campos de la histología, la biología celular y molecular, la clínica y la epidemiología; cuando dichos trabajos son el fruto de una continua dedicación de miles de científicos durante casi cien años; y cuando se sigue sin haber llegado todavía a una solución terapéutica para la enfermedad, nos damos cuenta de que nos encontramos ante un arduo problema médico.
Cuando pensamos además en la cifra de pacientes afectados (20 millones en los países desarrollados) y las características de la enfermedad (que origina una discapacidad permanente y que condiciona una dependencia total a un cuidador que, a su vez, ve su vida totalmente dependiente de la enfermedad), comprobamos que estamos ante un gran problema socio-sanitario a escala mundial que trasciende del ámbito de la medicina entendida en su concepto clásico.
Este es el caso de un terrible proceso neurodegenerativo, la enfermedad de Alzheimer (EA). No es de extrañar, por tanto, que todas las publicaciones científícas, clínicas, de divulgación socio-sanitaria e, incluso, de información general, estén llenas de artículos sobre esta demencia. Nos comunican nuevas aportaciones, nos analizan y reevaluan la ingente cantidad de datos generados en tantas investigaciones a través de los años o especulan con la aplicación de descubrimientos en otras patologías y en otros campos para resolver el problema Alzheimer. En este contexto todos los datos se entremezclan, a veces de manera lógica, pero otras no tanto. De ello surgen muchas veces propuestas de soluciones posibles, aunque también otras tantas se apunta hacia vías impracticables por falta de rigor científico en el análisis de esos datos o en su extrapolación al Alzheimer.
Bajo este prisma de sopesar ponderadamente los nuevos conocimientos sobre el Alzheimer y otras patologías degenerativas, y del crecimiento y la muerte celular, vamos a repasar brevemente las ciertamente posibles soluciones a esta demencia.
La complejidad de la etiopatogenia de la enfermedad de Alzheimer
Ciertamente pocas enfermedades como las neurodegenerativas presentan una tan abigarrada muestra de cambios celulares y moleculares y tan compleja relación entre los mismos. Como elementos «nucleares» del proceso han sido descritos cambios en proteínas fibrilares intraneuronales (que han dado lugar a las denominadas por algunos teorías tauístas, por el alto contenido de proteína tau en los ovillos neurofibrilares de las neuronas) o extraneuronales, manifestadas en los depósitos de amiloide (que danlugar a las denominadas teorías «baptistas» por un juego de palabras en inglés).
También son constantes las alteraciones en los sistemas colinérgicos basalocorticales (teorías colinérgicas), aumento neurotóxico de la transmisión excitatoria (teorías neurotóxicas), alteraciones en los sistemas neurotróficos y en la capacidad plástica y adaptativa de las neuronas, (teorías neurotróficas y adaptativas), aumento de los factores de envejecimiento y muerte neuronal programada, disfunciones gliales, etc.
Estos cambios están totalmente comprobados y las teorías a las que han dado lugar tienen total vigencia hasta que no se llegue a una incuestionable definición de la enfermedad, aunque precisan continuamente ser actualizadas por los nuevos conocimientos que surgen día a día.
Sin embargo, cualquiera de estos cambios, que es considerado «primario» o «fundamental» por los distintos autores que los utilizan cuando enuncian sus teorías sobre el Alzheimer (y que, por ello, implícitamente serían los objetivos de los tratamientos), tiene unas características comunes con importantes implicaciones en la enfermedad y su terapéutica:
- Ninguno de los cambios es patognomónico, es decir, todos ellos los podemos encontrar, en mayor o menor grado, en otras situaciones o patologías del Sistema Nervioso. Por ejemplo, el b-amiloide aparece, aunque en escasa densidad, en el envejecimiento y en otras patologías degenerativas o anóxicas.
- En un sentido muy estricto, los cambios son difíciles de precisar en sus límites, ya que existen elementos antecedentes, o consecuencias, muy interrelacionados que pudieran tener incluso más importancia que el cambio principal o más aparente. Por ejemplo, la reacción glial en la EA, descrita como «dramática» por algunos autores, no sólo no es específica de este proceso, sino que parece ser variable según las patologías, los individuos y las regiones cerebrales; estar condicionada por factores desencadenantes, muchos de ellos desconocidos, y producir efectos diferentes o cambiantes según dé lugar a la secreción de citoquinas, generalmente neurotóxicas, o de factores tróficos, generalmente neuroprotectores.
- Aunque algunos cambios tienen una conexión muy aparente entre sí y se pueden integrar con facilidad en una cadena de acontecimientos que pueden explicar la patogenia de la EA, muchos otros parecen estar desvinculados del proceso o, teóricamente, deberían estarlo. Por ejemplo, todavía no se han podido precisar una correlación satisfactoria entre la formación del b-amiloide extraneuronal de las placas y la de los complejos de proteína tau de los ovillos intraneuronales, aunque la coexistencia de estas dos lesiones fue ya descrita por el propio Alzheimer y ha sido estudiada hasta la extenuación. Por otro lado, aunque la amiloidosis y la gliosis se contemplan en algunas teorías como dos hechos íntimamente relacionados e inseparables, debemos desligarlos, ya que existen muchos procesos de gliosis aparéntemente similar en otras patologías sin presencia de amiloidosis.
- Todos los cambios se deben engarzar en una secuencia lógica, formando la denominada «cascada de acontecimientos patológicos». Pero, si difícil es definir los cambios y sus consecuencias, hasta ahora ha sido imposible describrir una «cascada» específica de la EA absolutamente comprobable, y así existen tantos o más modelos de cascadas como teorías sobre esta patología. Desde la situación inicial del cerebro óptimamente estructurado cumpliendo con sus funciones cognoscitivas al máximo nivel, hasta la situación final de un tejido dañado que es la base de toda la sintomatología de la demencia, ciertamente se han podido producir cambios secuenciales que por distintos caminos lleguen a la misma situación del deterioro. Sin embargo, parece más lógico pensar que causas pueden existir muchas (factores etiológicos), pero que la vía patogénica debe ser principalmente una con pocas variantes.
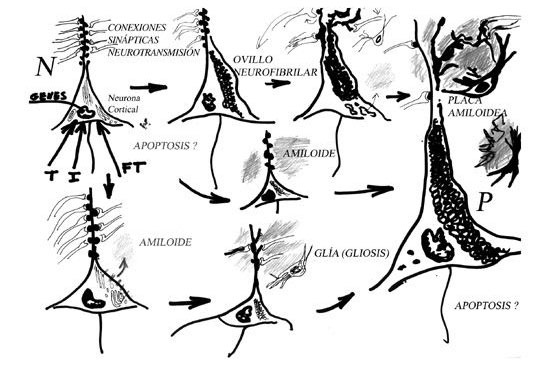
Disfunciones de los procesos fisiológicos celulares presentes en la patogenia de la EA y posibilidades terapéuticas
Entre las piezas que hay que encajar para tener una imagen de la cascada patogénica están una gran serie de cambios neuronales y gliales, algunos de los cuales (los más frecuentemente reseñados en la literatura) se recogen en la figura 1. Estos cambios comportan disfunciones de sistemas o procesos de las células que pueden manifestarse durante toda la vida celular o sólo en determinadas fases de la misma, desde el nacimiento y diferenciación hasta la muerte celular, pasando por distintas fases de adaptación y respuesta a estímulos, agresiones o cambios del medio ambiente (metabolismo, comunicación celular y neurotransmisión, adaptación celular, aprendizaje y memoria celulares, crecimiento y diferenciación, apoptosis, etc).
Estos sistemas o procesos se conocen principalmente por sus estudios en otras patologías, en modelos animales o celulares experimentales, o en ensayos in vitro. Estos estudios generan grandes conocimientos sobre los procesos, sus efectos, sus generadores fisiológicos y su patología. De ello podemos obtener la forma de regular su funcionamiento para que sea útil a la biomedicina.
Sin embargo, muchas veces es difícil, o por ahora imposible, extrapolar estos conocimientos al problema del Alzheimer, y menos sacar conclusiones prácticas, por la imposibilidad de encajar el proceso considerado en la «cascada patogénica», así como deslindar lo que es y significa este proceso en el conjunto de neuronas y células gliales cerebrales no afectadas.
Algunos procesos celulares presentes en la EA, que teóricamente parecen aberrantes y que, así mismo, con nuestros actuales conocimientos, parece que pueden ser corregidos, son las dianas terapéuticas sobre las que más se trabaja y se escribe, ofertándose como claves de soluciones seguras e inmediatas. Pero en la mayoría de los casos precisan una mayor investigación antes de entrar en ensayos clínicos por los problemas expuestos. En la monografía, trataremos especialmente estos procesos celulares como posible clave para interpretar y tratar la EA: la «muerte neuronal programada», el «mantenimiento y la involución de las neuronas» mediatizados por factores nerviosos y los «procesos inflamatorios» del cerebro. También se comentará en relación a estos procesos la posibilidad de desarrollar vacunas preventivas o curativas y las nuevas posibilidades de terapias con implantes celulares o con la inoculación de genes.
La muerte neuronal «programada»
Consideremos el caso de la apoptosis o muerte celular programada. Como resultado de gran número de investigaciones en diversos terrenos, se sabe que la célula viva normal posee unos sistemas (parcialmente funcionantes en toda su existencia, parcialmente inducidos ante distintos acontecimientos) que pueden desencadenar una serie de respuestas intracelulares que conducen a la autodestrucción. Principalmente están implicadas una serie de enzimas (caspasas, de las que se conoce hasta doce tipos) constitutivas o inducidas, que pasan de una forma inactiva a otra activa cuando se dispara el proceso de suicidio celular, y una serie de proteínas de una misma familia de las que existen elementos pro-apoptóticos y anti-apoptóticos (también muchas de ellas existentes en formas activas e inactivas y algunas que se interconvierten en uno u otro tipo). Muchas de estas enzimas y proteínas también tienen otras funciones no autodestructivas, pues intervienen en la regulación de la vida celular, estando implicadas en la realización de algunas funciones celulares de tipo adaptativo, especialmente en algunas circunstancias en las que cambia el medio interno o externo.
Pero, finalmente, si los cambios son «insuperables», y si la célula, pudiéramos decir, no encuentran otra salida a su crisis que sea menos perjudicial para el organismo, la maquinaria se pone en marcha hasta destruir completamente la célula. La apoptosis no es un fenómeno celular único sino que comprende varios procesos que pueden funcionar independiente o conjuntamente.
Quedan muchos puntos obscuros que aclarar, pero, ya existen en muchos laboratorios substancias de diverso tipo pro- y anti-apoptóticos para uso experimental que pueden desencadenar o detener la apoptosis.
En el Alzheimer, como en otras enfermedades neurodegenerativas, se ha postulado como agente causal una activación o descontrol de la apoptosis; ello ha sugerido la idoneidad de la aplicación de fármacos anti-apoptóticos en el tratamiento. Sin embargo, sabemos que la apoptosis comprende distintos procesos de autodestrucción, con diferentes desencadenantes y en la EA, el presunto proceso apoptótico y sus generadores inmediatos, son desconocidos. Así mismo, el objetivo final de la apoptósis en la EA nos es desconocido.
Cuando se analizan preparaciones de tejido cerebral de enfermos de EA, se observan, según la época de evolución de la enfermedad, la técnica que se emplee y la región que se considere, diversos signos de apoptosis cuya interpretación es discutible. ¿Tratan las neuronas que muestran estos signos de autoeliminarse para salvaguardar al tejido de problemas de neurotoxicidad y parar la «cascada patogénica», pero se ven impedidas a desaparecer, o, por el contrario, las neuronas que muestran estos signos son la imagen de que otros procesos patogénicos están matando muchas neuronas por la vía de un proceso de apoptosis? Es decir, ¿debemos favorecer la apoptosis para detener la progresión de la vía patogénica tisular, impidiendo que se fabriquen elementos tóxicos para otras neuronas, o, por el contrario, debemos detener la apoptosis para mantener con vida el suficiente número de neuronas para no interferir en los circuitos cognoscitivos y caer en la demencia?
Por otro lado, no parece posible, por el momento, diferenciar los sistemas apoptóticos de las neuronas en proceso de neurodegeneración, de los de las células normales que emplean, en el resto del organismo, estos sistemas apoptóticos para autoeliminarse (por su propia iniciativa o por señales que reciben de otras células) antes de ser dañinas, especialmente las que han sufrido trasformación cancerígena. Conocemos que los estrógenos favorecen el riego sanguíneo, comprometido en el Alzheimer, y que favorecen el balance de proteínas relacionadas con la apoptosis hacia la acción antiapoptótica.
Pero, un reciente estudio epidemiológico no ha mostrado efectos preventivos ni retardantes del curso clínico, mientras aumentaba el riesgo de cáncer de mama ya que impedía su diagnóstico precoz (JAMA, mayo). En consecuencia, por el momento, ni una medicación sistémica, ni una posible terapia intracerebral antiapoptóticas, podrían emplearse sin conocer en la EA el sentido y los mecanismos de la apoptosis.
Factores nerviosos de mantenimiento o de involución de las neuronas
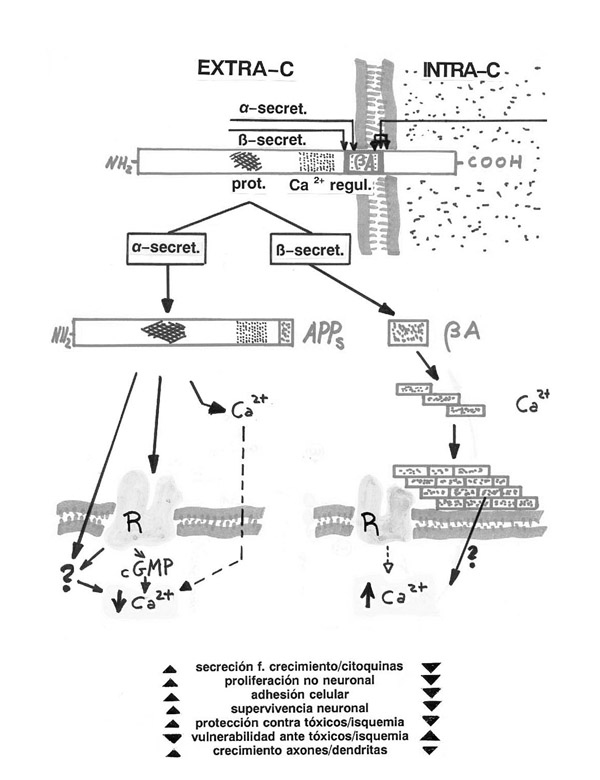
De una manera muy amplia, podríamos considerar que en el Sistema Nervioso Central (SNC) existen dos tipos de factores, producidos dentro del tejido, que, o bien mantienen vivas y plenamente funcionantes a las neuronas, especialmente las de regulación de la corteza cerebral como son las neuronas colinérgicas basalocorticales afectadas en la EA, denominadas neurotrofinas, o bien originan daños e involución neuronal, denominadas citoquinas. Sobre el papel que tienen en la EA estas importantes substancias, de igual forma que en el apartado anterior se podrían hacer consideraciones que cuestionan la idoneidad de su manipulación para tratar el proceso patológico. Una cosa es conocer con bastante precisión estas macromoléculas, sus sistemas producción, sus receptores y sus mecanismos iniciales de acción (neurotrófica o neurotóxica) y otra es saber cómo se regulan estos sistemas en la EA, qué receptores existen en cada neurona en condiciones normales de senilidad y en situación patológica, y cuáles son realmente los efectos finales que producen en las células. Hace pocos años se hacía equivalente el término de neurodegeneración con el de «déficit neurotrófico», habiéndose llegado a definir una teoría unitaria para todas las enfermedades neurodegenerativas (Alzheimer, Parkinson, Huntington, etc.) en base a la pérdida de factores neurotróficos (o a déficits de los sistemas neurotróficos) (Figura 2).
Esto actualmente es dudoso en su formulación original, e, incluso, en el inicio de la EA pueden existir fenómenos de hiperactividad de estos sistemas que, a posteriori, pueden volverse real o funcionalmente deficitarios. Se han llevado a cabo experiencias con infusiones intracerebrales de NGF (nerve growth factor, la neurotrofina por excelencia) en un humano que sufría EA, pero los resultados pusieron de manifiesto la inviabilidad de la terapia por lo costoso, por los problemas técnicos que surgieron y por los efectos secundarios graves (dolor, alteracionesconductuales, etc.). No es posible esta terapia si no conocemos bien los efectos sobre las neuronas no afectadas, si no sabemos con precisión la distribución de los receptores en las distintas regiones normales y alteradas del cerebro, … y si no tenemos la absoluta certeza de que no se van a activar receptores en células en vías de cancerización o de que no se van a dencadenar procesos tumorigénicos. En prácticamente todos los tipos de células cancerosas se han descrito altas densidades de receptores para neurotrofinas.
Respecto a las citoquinas, posiblemente neurotóxicas en la EA, cabría exponer que cada vez se sabe mejor que intervienen en muchos procesos fisiológicos neuronales y gliales, y en respuestas plásticas y adaptativas de las neuronas. Por ello, una terapia bloqueante de estas substancias debería analizar pormenorizadamente qué efectos tiene en distintos circuitos neuronales.
Procesos «inflamatorios» en el cerebro de enfermos de EA
La concepción del Alzheimer como un proceso inflamatorio, se viene contemplando de manera recurrente desde hace muchos años. Esto comporta la asimilación del conjunto de las estirpes gliales y sus substancias producidas (neurotrofinas, citoquinas) a las células y moléculas generadas del tejido conjuntivo, que existe en todo el organismo menos en el SNC, y que es el substrato de los fenómenos inflamatorios. En los últimos tiempos, se está haciendo especial hincapié en buscar soluciones para la EA en base a prácticas o técnicas que ya se aplican en el campo de la inmunología clínica, la alergología, la reumatología, etc, fundamentadas especialmente en el control de las reacciones inmunitarias y los procesos inflamatorios crónicos con vacunas y antiinflamatorios.
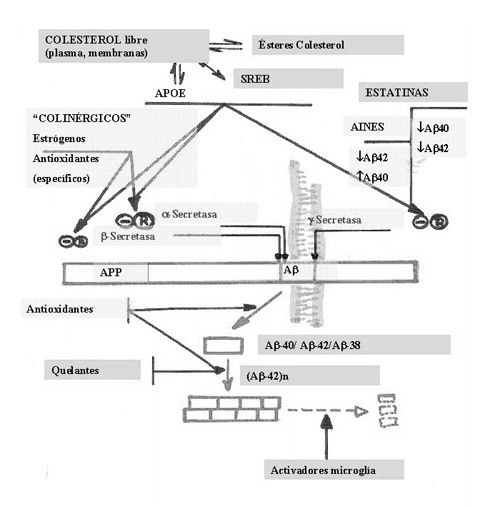
Las «vacunas» contra el Alzheimer han surgido tras la comprobación de que animales transgénicos con expresión del gen de la APP (proteína precursora de amiloide), presentaban disminución de la formación de placas en la senilidad cuando recibían anticuerpos contra una secuencia del péptido amiloide y manifestaban una cierta capacidad de disolución de las placas ya formadas cuando el tratamiento se iniciaba en época avanzada de la acumulación. Estos aparentes y llamativos efectos preventivos y curativos forzaron a la concesión de un permiso para experimentación clínica humana (sin seguir los trámites de experimentación preclínica) con dicho anticuerpo. Los ensayos fueron detenidos por fenómenos secundarios graves cerebrales en parte de los pacientes. Hace poco tiempo se publicó la autopsia del primer caso tratado y fallecido.
No se observaron diferencias grandes de conjunto con los enfermos de EA que no habían recibido vacuna y que se utilizaron como controles y sí en algunas zonas cerebrales parecía existir una tendencia a disminuir el número de placas con b-amiloide, en esas zonas parecía también que existían más ovillos neurofibrilares. La pérdida neuronal era similar en este enfermo y en controles pareados de EA que no recibieron el tratamiento. Difícil es sacar conclusiones de un ensayo, pero, aún con experiencias muy sobresalientes en modelos animales experimentales, sin conocer cual es el mecanismo, o el sistema o proceso, por el que se disminuye la acumulación de b-amiloide, no se puede extrapolar la función del sistema inmunitario basado en las células y propiedades del tejido conjuntivo a un órgano que no tiene este sistema. Por el contrario, parece más seguro y específico desarrollar fármacos capaces de regular las vías de metabolización de la APP para lograr que activando las a y g secretasas (vía «no amiloidogénica» de catabolización de la APP) o inhibiendo las b y g-secretasas (vía «amiloidogénica») se produzcan péptidos degradables y no b-amiloide (Figura 2). También pudiera ser viable, aunque por ahora no es practicable, que se llegara a activar la glía para destruir el posible amiloide formado. Esta forma de terapia sería de un alto interés ya que si se encuentra la manera de que las células puedan destruir las proteínas de configuración beta, se podrían combatir no sólo la EA sino también todas las enfermedades priónicas, patologías devastadoras del SNC, cuyos representantes en humanos (todas las variantes o subtipos de la enfermedad de Creutzfeldt-Jakob –esporádica; nueva variante, relacionada con el mal de las vacas locas; familiar o genética; yatrogénica, producida por transplantes o tratamientos hormonales–; el Imsomnio Familiar Fatal; la enfermedad de Gerstmann-Sträussler-Scheinker; el kuru) algunos autores piensan que pueden extenderse y aumentar su incidencia a valores muy preocupantes.
Respecto a la regulación de la «reacción inflamatoria» en la EA, se volvió a poner grandes esperanzas en los nuevos y potentes medicamentos anti-inflamatorios no esteroideos (AINES) que van incorporándose día a día en la práctica clínica, y que se basan en la inhibición de la ciclo-oxigenasa 2 (COX-2). Sin embargo, se acaba de publicar el último ensayo clínico (JAMA, junio 2003) donde se demuestra que ni se previene ni se mejora la EA con naproxeno o rafecoxilo, dos potentes anti-inflamatorios de nueva generación.
Esto puede ser decepcionante, pero otra vez se plantea el hecho de que no se conoce bien la implicación de los posibles sistemas pro-inflamatorios del cerebro y que emplean tanto la vía de la enzima ciclo-oxigenasa que, como se ha dicho, puede ser regulada, como por otras vías que utilizan la óxido nítrico sintasa y otras enzimas para producir substancias como prostaglandinas y óxido nítrico, que también son pro-inflamatorias. Además, si consideramos como reacción inflamatoria la destrucción y eliminación de células de un área dañada y reactiva, tenemos que incluir los fenómenos de reactividad glial que conducen a destrucción neuronal. En este sentido, en la patogenia de la EA es fundamental el proceso de activación anómala de las células gliales que producen unas substancias (citoquinas) que son citotóxicas en su mayoría, aunque algunas cumplen funciones fisiológicas aún no bien conocidas. Quizás la terapia anti-inflamatoria más eficaz sería aquella que regulara la producción de citoquinas, de manera que no se produjera toxicidad neuronal pero que mantuviera los niveles de estas substancias cumpliendo con su papel fisiológico.
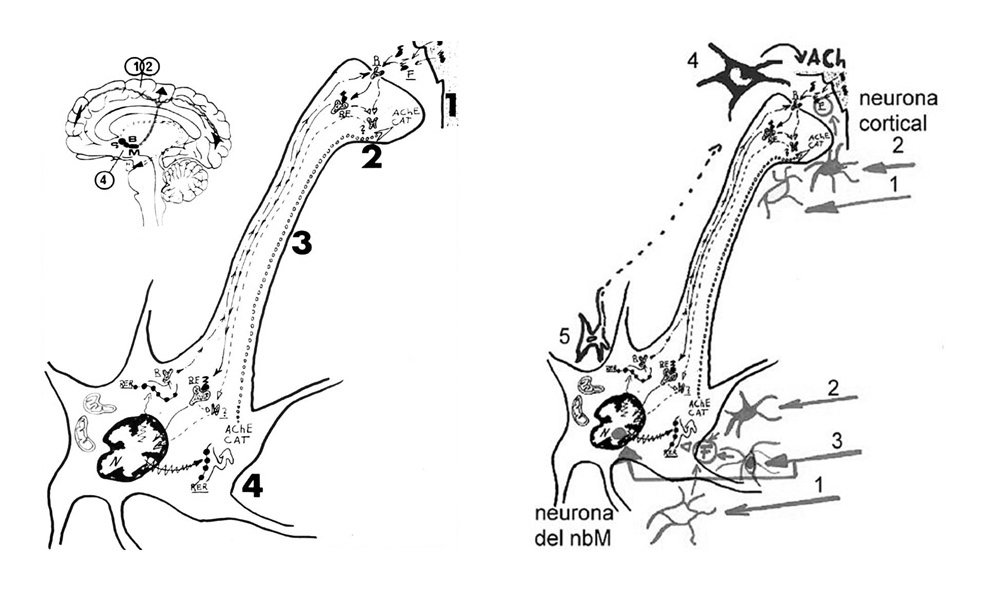
Sistemas neurotransmisores en EA
Uno de los fenómenos más importantes en la patogenia de la EA es la disminución de los sistemas de neurotransmisión reguladores. En especial, el sistema colinérgico de todo un conjunto amplio de neuronas situadas en la parte basal del cerebro y que alcanza prácticamente a todas las neuronas de las distintas áreas de la corteza cerebral que están implicadas en la realización de las funciones congnoscitivas superiores del cerebro. Los estudios sobre los sistemas neurotransmisores in vitro o en modelos experimentales nos proporcionan informaciones muy precisas sobre cada uno de los tipos y subtipos en todos sus elementos (enzimas, receptores, segundos mensajeros
intracelulares). Nuestro conocimiento es muy amplio y ya tenemos fármacos para modificar cualquier sistema en el sentido deseado, pero los resultados clínicos no son buenos ya que existen problemas importantes en la extrapolación de los estudios. Para tener éxito se debería actuar de forma selectiva sobre las neuronas o circuitos alterados y corrigiendo exactamente la alteración que existe en esa localización. Muchas células del organismo poseen receptores similares y una terapia sistémica general podría provocar graves efectos colaterales. Tanto en el aspecto de los sistemas deficitarios como el colinérgico, como en el de los presuntamente hiperactivos (nueva teoría de la «neurotoxicidad glutamatérgica» que se basa en una supuesta hiperactividad de las neuronas excitadoras de la corteza cerebral que hace que se dañen o mueran las neuronas reguladoras del cerebro basal, especialmente las colinérgicas), hace falta, por un lado, precisar con exactitud los tipos y subtipos de receptores normales y patológicos que cada circuito normal y patológico poseen, y en segundo lugar, obtener fármacos específicos de los receptores patológicos y que no interfieran con las funciones de las células normales. Si empleamos un fármaco colinérgico de tipo nicotinérgico ciertamente mejoraríamos las funciones corticales, pero con el riesgo de provocar importantes problemas cardiovasculares.
Por otra parte, en muchos casos, la función que estamos recuperando con la terapia dista mucho de ser la fisiológica que se ha deteriorado o perdido. Pensemos, por ejemplo, que fisiológicamente la acetil-colina actúa en la corteza cerebral durante milisegundos tras su liberación por los terminales de las neuronas colinérgicas de los núcleos basalocorticales, pues es rápidamente metabolizada por la enzima acetil colinesterasa, y que, en cambio, los anticolinesterásicos empleados en la EA para suplir el déficit de acetilcolina pueden conducir a una actuación de minutos a horas de este neurotrasmisor sobre las neuronas corticales. Esto, además, puede tener un efecto secundario añadido de disminución de síntesis de acetilcolina por retroalimentación negativa. Para obviar todos estos inconvenientes ya se están desarrollando algunos fármacos selectivos de subtipos de receptores colinérgicos nicotínicos específicos de la corteza cerebral. Ahora habría que hacer lo mismo con los subtipos de receptores farmacológicamente distinguibles por su afinidad con el N-metil-D-apartato (NMDA) de receptores para glutamato, ya que fármacos como la memantina, que se empleaba en el tratamiento de algunas patologías del SNC, se ha autorizado en la clínica sin mayores ensayos previos y, a consecuencia de ello, aunque ciertos estudios están mostrando algunos beneficios, los resultados son discutibles para muchos investigadores, al mismo tiempo que se piensa que se están propiciando muchas alteraciones, ya que se interfieren algunas funciones normales de las neuronas.
En el campo futurista del implante de células en el cerebro con fines terapéuticos (dejando aparte el problema ético de las células madre y pensando en el desarrollo de la aplicación de las células madre adultas, Acta Científica y Tecnológica, 5:31-35, 2002), así como el de la terapia génica, existen muchas posibilidades en un futuro cercano, pero también vías ciertamente de imposible desarrollo (Figura 3). Podrían obtenerse, y ya se está consiguiendo, neuronas secretoras de neurotransmisores deficitarios (como acetilcolina) y neuronas o células gliales productoras de neurotrofinas (NGF, otras neurotrofinas) que implantadas en lugares estratégicos (como diversas regiones de la corteza o núcleos basales del cerebro), compensaran los déficits basales regionales y pudieran también responder a estímulos fisiológicos cuando fuera necesario. En un sentido similar, podrían las células remanentes hipofuncionantes ser «rehabilitadas» por la inclusión de genes (vehiculizados o no por virus) productores de las substancias deficitarias (neurotransmisores, substancias neurotróficas, receptores) o de elementos que las produzcan (enzimas).
Sin embargo, debemos considerar una vía imposible hoy en día (y por muchos años) las substitución de neuronas que pudieran llegar a configurar nuevos circuitos cognoscitivos que substituyeran a los perdidos o deteriorados. La absoluta complejidad de los circuitos con sus conexiones colaterales, las miles de millones de neuronas implicadas en cada una de las funciones cognoscitivas y la comprobación de que aun conservando la especialización de cada circuito, el cerebro funciona como un todo, hacen de imposible cumplimiento el aserto de que las enfermedades neurodegenerativas tendrán su fin con el desarrollo de neuronas a partir de células madre embrionarias, aunque intereses no confesables machaconamente insistan sobre esta «posibilidad».
Agradecimiento
Agradezco a la Dra. María Isabel Álvarez su valiosa y decidida colaboración en la redacción de la monografía así como en los estudios sobre la Enfermedad de Alzheimer.
Conclusión
El conocimiento de la Enfermedad de Alzheimer, así como el desarrollo de terapias preventivas, paliativas o curativas, viene tanto de la investigación de casos de EA y sus modelos experimentales como la de los procesos o sistemas afectados realizada en otras patologías, y sus modelos in vitro. Estos últimos estudios parecen aportar mayores conocimientos y apuntar muchas mejores soluciones, pero su extrapolación al Alzheimer resulta muy problemática y da lugar al anuncio de posibles tratamientos que son imposibles tanto teórica como practicamente. Todos nuestros conocimientos generales debidos al avance de la biomedicina deben ser específicamente reconsiderados en la EA antes de lanzar al mundo soluciones para este terrible problema y no crear estados de grandes esperanzas y grandes decepciones.
Bibliografía
- Aisen PS, y cols. Alzheimer’s Disease Cooperative Study. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 289:2819-2826 (2003).
- Auld DS, y cols. Alzheimer’s disease and the basal forebrain cholinergic system:relations to beta-amyloid peptides, cognition, and treatment strategies. Prog Neurobiol 68:209-45 (2002).
- Clark CM, Karlawish JH. Alzheimer disease: current concepts and emerging diagnostic and therapeutic strategies. Ann Intern Med 138:400-410 (2003).
- Conway KA, y cols. Emerging beta-amyloid therapies for the treatment of Alzheimer’sdisease. Curr Pharm Des 9:427-47 (2003).
- De Felice FG, Ferreira ST. Beta-amyloid production, aggregation, and clearance as targets for therapy in Alzheimer’s disease. Cell Mol Neurobiol 22:545-63 (2002).
- DeKosky ST. Pathology and pathways of Alzheimer’s disease with an update on new developments in treatment. J Am Geriatr Soc. 51(Sup 2):S314-20 (2003).
- Giacobini E. Cholinesterases: new roles in brain function and in Alzheimer’s disease. Neurochem Res 28:515-522 (2003).
- Hardy J. The Relationship between Amyloid and Tau. J Mol Neurosci. 20:203-6 (2003).
- Hartig W, y cols. Functional recovery of cholinergic basal forebrain neurons under disease conditions: old problems, new solutions?. Rev Neurosci 13:95-165 (2002).
- Janus C. Vaccines for Alzheimer’s disease : how close are we?. CNS Drugs 17:457-474 (2003).
- Kamenetz F, y cols. APP processing and synaptic function. Neuron. 37:925-937 (2003).
- Marks N, Berg MJ. APP processing enzymes (secretases) as therapeutic targets: insights from the use of transgenics (Tgs) and transfected cells. Neurochem Res 28:1049-1062 (2003).
- Mattson M. Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromolecular Med 3:65-94 (2003).
- McGeer PL, McGeer E. Is there a future for vaccination as a treatment for Alzheimer’s disease?. Neurobiol Aging 24:391-395 (2003).
- Nagele RG, y cols. Astrocytes accumulate Abeta42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res 971:197-209 (2003).
- Nicoll JA, y cols. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med 9:448-52 (2003).
- Olney JW, y cols. Glumate Receptor Dysfunction and Alzheimer’s Disease. RestorNeurol Neurosci 13:75-83 (1998).