06 Feb 2014 La enfermedad de Alzheimer: bases moleculares y aproximaciones terapéuticas
Ana Castro y Ana Martínez
Instituto de Química Médica, Centro Superior de Investigaciones Científicas (CSIC)
Introducción
La enfermedad de Alzheimer (EA) recibe su nombre debido a las contribuciones científicas de Alois Alzheimer sobre un tipo de desorden neuropsiquiátrico. Las investigaciones de Alzheimer se basaron en establecer una correlación entre la sintomatología clínica de pacientes con este desorden, y la aparición de estructuras anómalas en el cerebro de los mismos.
En su trabajo experimental a comienzos del siglo pasado, Alois Alzheimer observó mediante tinciones con plata de fragmentos de cerebro provenientes de autopsias de tales enfermos, la presencia de ovillos neurofibrilares. Estas estructuras fueron encontradas en el cerebro de pacientes que experimentaban un deterioro progresivo en sus funciones cognitivas y en su memoria, hasta llegar a un estado en que prácticamente no recordaban nada. Sin embargo, no se encontraban presentes en los cerebros post-mortem de individuos controles mayores de edad que no tenían tal dolencia.
Hoy en día, la EA representa la forma más común de demencia en personas adultas. La incidencia aumenta con la vida media de la población afectando entre el 47 y 50% de los mayores de 85 años. En las últimas décadas, la población mundial ha sufrido importantes cambios en su estructura por edades, sobre todo en los países desarrollados.
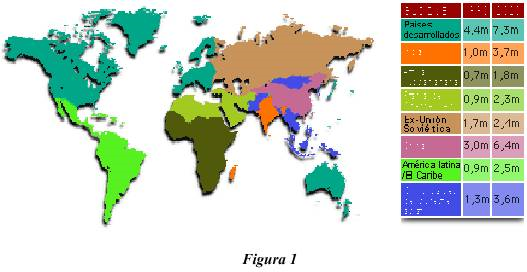
Así, y debido al progresivo envejecimiento de la población mundial y al aumento creciente de la esperanza de vida media se estima que, en el año 2020, tan solo en los países desarrollados, en torno a los siete millones de personas se vean afectadas por esta enfermedad (figura 1).
Por todo ello, el impacto, no solo social, sino también económico, que esta enfermedad está suponiendo para nuestra sociedad es muy grande.
Presentación clínica y características neuropatológicas
La EA es un proceso neurodegenerativo múltiple del sistema nervioso central, que se caracteriza clínicamente por la pérdida progresiva de la memoria a corto plazo y de la atención, seguida de la afectación de otras habilidades cognitivas, como el lenguaje y el pensamiento abstracto, el juicio crítico y el reconocimiento de lugares o personas.
En las primeras fases de la enfermedad, el impacto psicológico en el paciente es devastador, y en estadios avanzados, éste evoluciona a un mutismo casi absoluto con un deterioro progresivo de sus capacidades motrices pudiendo llegar a una total desconexión con el entorno, siendo incapaz de controlar sus funciones fisiológicas más simples.
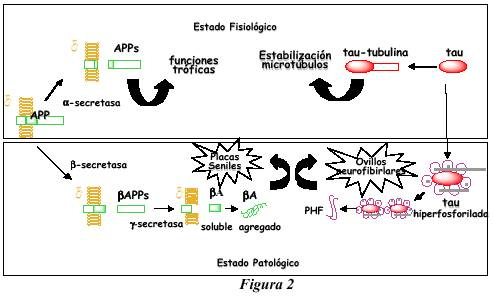
A nivel histopatológico, la EA se asocia a la formación masiva de dos tipos de agregados protéicos: los ovillos neurofibrilares que se localizan en el interior de la neurona y las placas seniles en el espacio extracelular (figura 2).
Los ovillos neurofibrilares están formados por los filamentos pareados helicoidales (PHFs), estructuras anómalas de la neurona, cuya presencia provoca serios trastornos en la actividad de ésta que la llevan a una pérdida de su capacidad de transmitir mensajes nerviosos, y finalmente al proceso neurodegenerativo. Se sabe que las neuronas que contienen ovillos neurofibrilares pierden su capacidad funcional, y muchas de ellas mueren como se evidencia por la presencia de residuos neuronales que contienen dichos ovillos en el cerebro de pacientes con la enfermedad de Alzheimer.
Por otra parte, las placas seniles son estructuras esféricas localizadas en el espacio extracelular donde desplazan las terminaciones nerviosas. Se trata de conglomerados anulares de cuerpos y prolongaciones neuronales degeneradas en torno a un depósito central de un péptido de longitud variable (de 40 o 42 amino ácidos) llamado -amiloide (A).
El A depende de la ruptura enzimática de la proteína precursora de amiloide (APP).Tres enzimas son responsables de este proceso de ruptura. APP puede fragmentarse por acción de la -secretasa, seguida de la acción de la -secretasa. De manera que se genera fragmentos solubles de APP. Sin embargo, cuando sobre APP actúa en primer lugar las -secretasa seguido de la acción de -secretasa se libera los fragmentos de A (1-40)y A (1-42),poniéndose en marcha la ruta amiloidogénica.
Los ovillos neurofibrilares constituyen la principal lesión intraneuronal y se encuentran fundamentalmente en los cuerpos neuronales y dendritas apicales, aunque en menor proporción se detectan lesiones neurofibrilares también formadas por PHFs en dendritas distales como los filamentos de la neuropila y en neuritas distróficas que rodean los núcleos centrales de algunas placas del amiloide. Dichas estructuras anómalas de la neurona se forman principalmente a partir de ta , una proteína asociada a los filamentos que forman el citoesqueleto neuronal.
En condiciones normales de la neurona, la proteína tau juega un papel fundamental en la modulación de la formación de los microtúbulos, polímeros claves de la arquitectura neuronal y que son además esenciales para mantener la dinámica del citoplasma, procesos de transporte en el interior de la neurona y en la formación del huso mitótico en células en división.
Debido a una alteración de las señales regulatorias, tau deja de cumplir su papel en el mantenimiento de la estabilidad del citoesqueleto y se transforma en una proteína con una capacidad aberrante de asociarse consigo misma para formar polímeros intracelulares. Dichos polímeros de tau son los que se organizan en las estructuras helicoidales altamente resistentes de los PHFs.
Por otro lado, durante el proceso de degeneración neurofibrilar que caracteriza esta enfermedad se produce una perdida notable de la inervación colinérgica de la corteza cerebral, sobre todo en el hipocampo y en el neocortex. Se ha encontrado una disminución importante en los niveles de ciertos neurotrasmisores. Concretamente, se ha notado una reducción considerable de los niveles de acetilcolina y disminución de la colinacetiltransferasa, lo que se ha relacionado con pérdida de neuronas colinérgicas en regiones del cerebro que están implicados en los procesos de memoria y aprendizaje.
Además, se ha observado déficits en otros sistemas como el noradrenérgico, dopaminérgico y serotoninérgico.
Patogénesis
La investigación, a lo largo de los últimos años, acerca de la EA ha sido muy intensa y como resultado de ella se están postulado un gran número de hipótesis que ayudan a entender cada día más este complejo proceso neurodegenerarivo. Si bien hoy en día no se conoce la etiología de esta enfermedad todos los datos acumulados a lo largo de las últimas décadas apuntan a un conocimiento del origen la misma cada vez más próximo.
A continuación, se recogen algunas de las hipótesis más representativas sobre las que se ha centrado fundamentalmente la investigación en esta área.
Hipótesis colinérgica
De todas las hipótesis que intentan describir la patogénesis de la EA, la que más se ha estudiado y probado ha sido probablemente la llamada hipótesis colinérgica. Con datos que van desde experimentos a nivel molecular hasta ensayos clínicos basados en terapias colinérgicas. De hecho, los únicos tratamientos farmacológicos que actualmente se administran para la mejora cognitiva de los pacientes son fármacos colinérgicos.
La correlación encontrada entre el déficit colinérgico (entre otros, disminución del neurotransmisor acetilcolina) y la perdida de las capacidades cognitivas de los enfermos es lo que ha motivado la intensa investigación en este punto, siendo una de las aproximaciones terapéuticas más explotada y desarrollada en los últimos años. Como resultado de la misma ha sido la puesta en el mercado los únicos fármacos comercializados hasta el momento para el tratamiento paliativo de esta enfermedad.
Basándose en la estructura y los mecanismos bioquímicos de la sinapsis se están desarrollado las siguientes estrategias terapéuticas: precursores de acetilcolina, agonistas nicotínicos, liberadores de neurotransmisores, agonistas muscarínicos M1 o antagonistas muscarínicos M2 y inhibidores de acetilcolinesterasa.
Concretamente, los fármacos actualmente comercializados actúan por este último mecanismo. Tal es el caso de tacrina (Cognex), donepezilo (Aricept )o rivastigmina (Exelon).
Hipótesis excitotóxica
El glutamato es el principal neurotransmisor excitatorio y de hecho el sistema glutamatérgico está implicado en los acontecimientos excitotóxicos que tienen lugar en otras muchas patologías neurodegenerativas, tales como isquemia, esclerosis lateral amiotrófica, esclerosis múltiple entre otras.
De acuerdo con la teoría excitotóxica la activación glutamatérgica de los receptores NMDA y/o AMPA/kainato, conduce a una entrada masiva de calcio. Como resultado del aumento desmesurado de las concentraciones de calcio intraneuronal se inducen la activación de gran variedad de compuestos neurotóxicos que alteran y disminuyen la viabilidad neuronal,conduciendo en definitiva a la muerte neuronal.
Las aproximaciones terapéuticas que se están teniendo en cuenta se centran en el diseño de fármacos neuroprotectores, entendiendo por tales todas aquellas sustancias que disminuyen en definitiva la liberación de los aminoacidos excitatorios controlando de esa manera sus efectos intracelulares. Así se pueden considerar como tales el desarrollo de antagonistas del receptor NMDA, antagonistas del receptor AMPA, bloqueadores de canales de calcio, inhibidores de la sintasa del óxido nítrico, captadores de radicales libres, antagonistas de los canales de sodio, inhibidores de la liberación de glutamato o activadores de canales de potasio.
Daño oxidativo y procesos neuro-inflamatorios
Otra de las teorías que intentan explicar la patogénesis de la EA es la teoría de los radicales libres. El daño producido por el exceso de las especies radicales es algo asociado a los procesos de envejecimiento y en la EA el daño oxidativo juega un papel importante ya que los radicales libres atacan a las neuronas produciendo la oxidación de lípidos, proteínas y ADN, lo que se traduce en la muerte neuronal.
En condiciones normales, las especies radicalicas son controladas por una eficiente cascada de mecanismos de antioxidación, que incluye tanto la intervención de diferentes enzimas antioxidantes como de agentes antioxidantes no-enzimaticos.
Sin embargo, durante los procesos neurodegenerativos se produce una descompesanción entre la producción de los radicales libres y la defensa antioxidante celular, como consecuencia se producen fallos en diferentes funciones biológicas conduciendo a la muerte celular.
Por otro lado, en cerebros con Alzheimer se producen un gran número de factores neuro-inflamatorios como son: inmunoproteínas y citoquinas generadas por neuronas, astrocitos y microglia. Además de la generación de especies radicalicas, contribuyendo a agravar las situaciones de daño oxidativo. De manera que el daño oxidativo y la cascada neuro-inflamatoria contribuyen de forma paralela a la patogénesis de esta enfermedad ofreciendo nuevas estrategias para el desarrollo de fármacos.
De acuerdo con el punto de intervención farmacológico, se podrían clasificar como: antioxidantes, captadores de radicales libres o anti-inflamatorios neuronales.
Proteína tau
Otro de los marcadores característicos de la EA es la presencia de ovillos neurofibrilares, De hecho, la correlación entre la presencia de estos y el grado de demencia es uno de los indicadores en la EA.
Uno de los componentes de los ovillos neurofibrilares es la proteína tau. Como se ha comentado anteriormente, la función celular de la misma es modular la adhesión de los microtúbulos y su estabilización en las neuronas.
Una de las causas probables por las cuales se produce un cambio en la conducta funcional de tau sería la fosforilación anormal en sitios críticos de su estructura, disminuyendo su capacidad para promover la adhesión de los microtúbulos bloqueando su acción estabilizadora.
El mecanismo de fosforilación de la proteína tau in vivo es todavía incierto, aunque son dos las quinasas más relevantes conocidas: la denominada TPK I, que es idéntica a la GSK-3 y la TPK II formada por un nuevo activador de la proteína p35 y una unidad catalítica que resulta ser idéntica a la quinasa dependiente de ciclina 5. De manera que la inhibición de estas enzimas constituye un objetivo terapéutico de alto interés en la búsqueda de fármacos anti-Alzheimer.
B-amiloide
La presencia de placas extracelulares de BA es un hecho central en la patología de la EA. La teoría del B-amiloide se fundamenta en que los agregados de BA son el factor desencadenante de multitud vías neurotóxicas, entre las que se pueden incluir exicitotoxicidad, alteraciones en la homeostasis del calcio, producción masiva de radicales libres y procesos neuro-inflamatorios. Las estrategias terapéuticas van desde los inhibidores de B y Y secretasas, enzimas responsables de la fragmentación anómala de la proteína precursora del amiloide, hasta la búsqueda de antiagregantes de dicho péptido.
Sin embargo, hoy en día no existe consenso acerca de cómo la deposición del amiloide lleva a la demencia y si los depósitos de BA son suficientes por si sólo para causar la enfermedad. Fundamentalmente debido a que los estudios de Alzheimer ya indicaban que las placas seniles se encontraban tanto en cerebros con la enfermedad como en los controles siempre que se tratara de ancianos, lo que sugería que tales placas podrían ser marcadores de senilidad más que de demencia.
En apoyo de esto, existe evidencia que la distrofia neurítica se correlaciona con la expresión de formas de demencia clínica, y que los pacientes pueden tolerar ciertos niveles de amiloidosis antes de presentar signos de disturbios cognitivos. La formación de componentes del amiloide es común en el envejecimiento normal, y en casos muy raros se encuentran ovillos sin la presencia de amiloide.
De esta forma, el BA posiblemente precedería a la ocurrencia de ovillos neurofibrilares. Sin embargo, es posible que los dos eventos celulares claves en el Alzheimer, formación de PHFs y de placas seniles, lleven en forma complementaria a la pérdida de la actividad de las neuronas afectadas. Aunque son fenómenos molecularmente independientes, la generación de PHFs y la deposición del amiloide, pudiesen tener alguna relación a nivel del funcionamiento neuronal, aunque hoy en día este sigue siendo un debate abierto entre los baptistas ç (defensores del B-amiloide como agente etiológico primario causante de la EA) y los tauistas (defensores de la disfunción de la proteína tau como agente etiológico primario causante de la EA).
Conclusiones
En el estado actual de la investigación biomédica, aún no existe un tratamiento para la cura de la enfermedad de Alzheimer cuya eficacia esté reconocidamente demostrada, y los enfoques terapéuticos están orientados a obtener un cierto grado de mejoría del paciente en etapas iniciales de la enfermedad.
La búsqueda de un tratamiento eficaz contra la enfermedad de Alzheimer dependerá del mayor conocimiento de sus mecanismos biológicos. Sin embargo, la neurobiología del Alzheimer es muy compleja.
Como ya se ha comentado, uno de los cambios importantes a nivel celular en la enfermedad de Alzheimer es la pérdida gradual de neuronas de los núcleos basales, cuyo neurotrasmisor es la acetilcolina, y cuyas prolongaciones llegan a la corteza cerebral. Para controlar en parte éste y otros efectos se han usado múltiples medicamentos entre los cuales están tacrina (Cognex), donepezilo (Aricept) o rivastigmina (Exelon), que se manejan actualmente con fines terapéuticos.
La formación de B-amiloide así como la de los PHFs son blancos potenciales para diseñar aproximaciones terapéuticas contra el Alzheimer, y ello dependerá del conocimiento que se tenga sobre los mecanismos moleculares de la generación de estas estructuras.
No hay que olvidar, como vía de investigación terapéutica el bloqueo de enzimas que cortan en forma anómala la proteína amiloide, mediante el diseño de fármacos que desactiven las enzimas aberrantes que conducen a la formación de las placas, permitiendo así la fragmentación normal del componente amiloide. Asimismo, se podrían controlar selectivamente los mecanismos enzimáticos que determinan las hiperfosforilaciones en tau y su agregación en PHFs.
En general, todas aquellas vías que permitan controlar los procesos bioquímicos alterados en la EA están proporcionando nuevos puntos de intervención farmacológica que, unido al desarrollo de los animales transgénicos, como método de evaluación de fármacos, están abriendo las puertas hacia posibles enfoques farmacológicos que aseguran un halo de esperanza en la búsqueda de un futuro tratamiento para esta enfermedad.
Referencias
- Wiltfang, J.; Esselmann, H.; Maler, J.M.; Bleich, S.; Huther, G.; Kornhuber, J. Molecular biology of Alzheimer ’s disease dementia and its clinical relevance to early diagnosis and new therapeutic strategies. Gerontology, 2001, 47, 65-71.
- Hirai, S. Alzheimer’s disease: current therapy and future tharapeutic strategies. Alzh. Dis. Assoc. Disord., 2000 ,14, S11-S17.
- Grundman, M.; Thal, L.J. Treatment of Alzheimer ’s disease: rationale and strategies. Neurol. Clin., 2000, 18, 807-828.
- Smith, R.G. The aging process. Where are the drug opportunities? Curr.Opin.Chem.Biol., 2000, 4, 371-376.
- Gauthier, S. Alzheimer’s disease: current and future therapeutic perspectives. Prog. Neuropsychopharmacol. Biol. Psychiatry, 2001, 25, 74-89.
- Latimer, L.H. Therapeutic approaches to Alzheimer’s disease.Curr. Opin. Chem. Biol., 2000, 4, 377-382.
- Qizilbash, N.;Emre, M. Experimental approaches and drugs in development for the treatment of Alzheimer’s disease.Expert Opin. Investig. Drugs, 2001, 10, 607-617.
- Cutler, N.R.; Sramek, J.J. Review of the next geneartion of Alzheimer ’s disease therapeutics: challenges for drug development. Prog. Neuropsychopharmacol. Biol. Psychiatry., 2001, 25, 27-57.
- Schneider, L.S. Treatment of Alzheimer ’s disease with cholinesterase inhibitors. Clin. Geriatr. Med., 2001, 17, 337-358.
- Tsopelas, N.D.; Marin, D.B. Cholinergic treatments of Alzheimer’s disease. Funct. Neurobiol. Aging, 2001, 475-486.
- Castro, A.; Conde S.; Rodriguez-Franco, M.I.; Martinez, A. Non-Cholinergic Pharmacotherapy to the Future Treatment of Alzheimer’s disease,Mini-Rev.Med.Chem. 2002, 2,37-50.