24 Nov 2016 ¿Qué son las enfermedades raras?
Luisa-María Botella
Centro de investigaciones biológicas. CSIC. Madrid.
Son enfermedades poco frecuentes en la población. No es que los pacientes sean raros, sino que la rareza viene definida por la escasez de personas que padecen cada una de estas patologías. En Europa se ha definido a una patología como rara cuando afecta a menos de cinco personas cada 10.000 habitantes (equivale a menos de uno cada 2.000). En EEUU se define como rara aquella que afecta a menos de 200.000 personas dentro del territorio de los Estados Unidos de América. El número de afectados por enfermedades raras varía desde las menos raras (1:2000-5.000), más raras (1:36.000-50.000), muchas extremadamente raras (que las padecen sólo una de cada 100.000 personas), incluso otras ultra-raras (dos o tres afectados en España) y muy pocas personas en la Unión Europea.

«He conocido enfermedades tan raras, que podría decirse que es en el paciente donde se expresa por primera vez.»
Maimónides: médico y teólogo judío de la cultura de Al-Andalus (1135-1204)
Existen entre 5.000 y 7.000 enfermedades raras en total. Por tanto en conjunto podemos decir que no es tan raro tener una enfermedad rara. Hay unos 30 millones de personas afectados por una u otra enfermedad rara en los 25 países europeos, lo que representa del 6% al 8% de la población mundial.
No es tan raro tener una enfermedad rara, en España somos unos tres millones de personas afectadas.
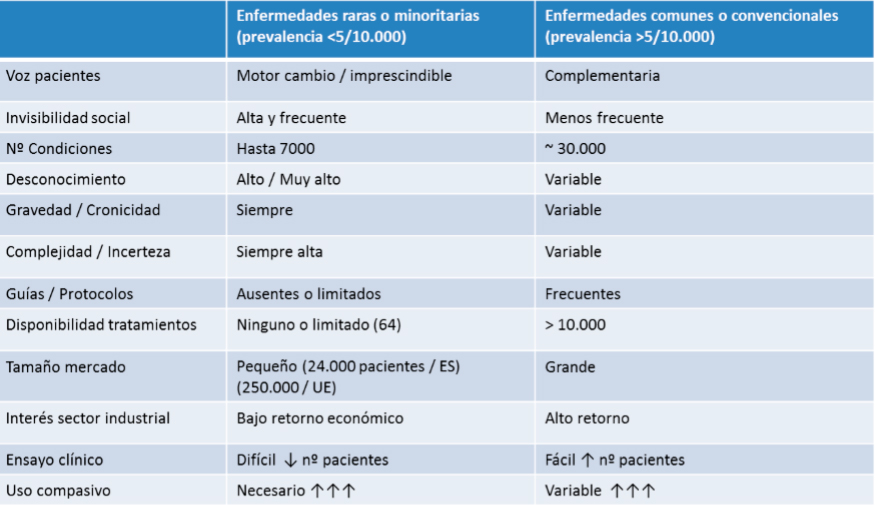
Características comunes a las enfermedades raras o poco frecuentes (ER o EPF)
El pronóstico vital está en juego en el 50% de los casos. A las Enfermedades Raras se les puede atribuir el 35% de las muertes de niños y niñas menores de un año y el 10% de mortalidad infantil entre uno y cinco años. El 30% de los pacientes fallece antes de los cinco años. El 50% fallece antes de los 30 años. No existe cura para la mayoría de las ER. El 80% son de origen genético.
Tabla. Características comunes a las enfermedades raras o poco frecuentes (ER o EPF)
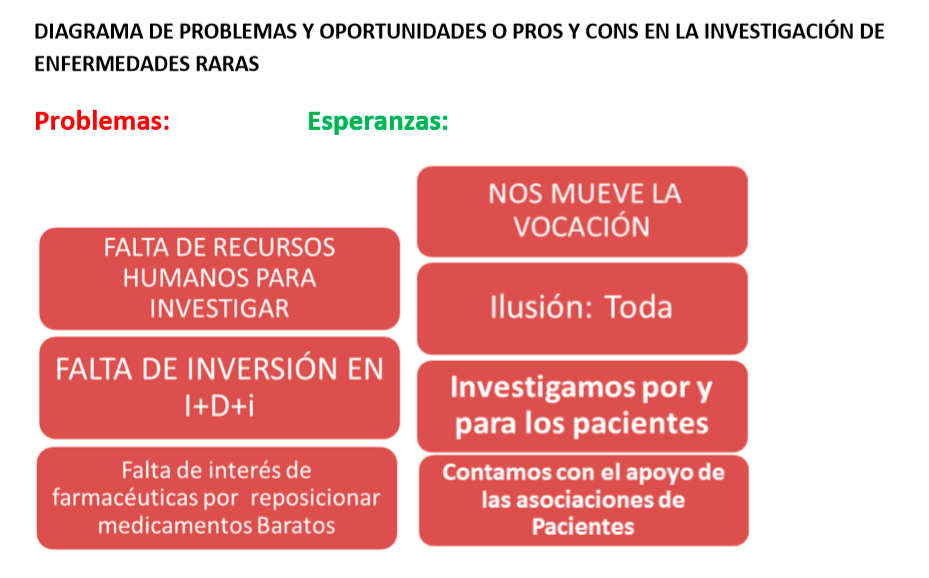
La realidad, dificultades comunes a todas las enfermedades
Como cada enfermedad rara individualmente representa a una minoría de pacientes, no son una prioridad para las políticas de Salud Pública. Sólo en los últimos años se están empezando a tener en cuenta, gracias a la visibilidad y difusión de este tipo de patologías por parte de las asociaciones de pacientes y familiares.
Se realiza poca o ninguna investigación. La industria farmacéutica no invierte dinero en investigación de tratamientos cuyas ventas futuras no llegarán a cubrir el dinero necesario para desarrollar la investigación.
Desde diferentes gobiernos y asociaciones de pacientes han planteado ayudas económicas en forma de incentivos, para animar a la empresa farmacéutica a desarrollar medicinas destinadas a enfermedades raras “huérfanas” del interés económico de otras enfermedades. Por ello para enfermedades raras, los medicamentos desarrollados se han denominado, “medicamentos huérfanos”.
¿Quién sufre la enfermedad?
En primer lugar, quien la padece en su propio cuerpo. Sin embargo, no es el único afectado. En realidad toda la familia está afectada, aunque los demás miembros no tengan la enfermedad. La influencia de la enfermedad es tan grande, que toda la familia se considera afectada psicológicamente, emocionalmente y a veces con cargas de dependencia del afectado que recaen sobre uno de los miembros de la familia, a menudo la madre.
Todas las personas que nos quieren, nuestros amigos y nuestros compañeros, nuestro entorno social. Todos sufrimos las consecuencias.
¿Cómo se siente una persona con una enfermedad así?
A continuación reproducimos la voz de algunos afectados:
− Los médicos no conocen mi enfermedad… Eres tú realmente quien va informando a los médicos…
− Te sientes como un bicho raro.
− Sientes miedo porque piensas: “si ellos, que son los médicos, no la conocen, ¿cómo me van a ayudar a mí y a mi familia?”
− No hay nada para solucionarlo, no hay tratamiento. Te dicen “tienes esto, pero no hay nada que podamos hacer”.
− Los demás no entienden mi problema: a veces me miran otros niños por la calle, y se ríen de mí. Sufro mucho por esto.
− Hay que recordar continuamente a los profesores que hay determinadas cosas que los niños no pueden hacer.
− No puedo ir al cine ni a otros espectáculos, porque donde yo vivo no hay salas adaptadas para sillas de ruedas.
− Así es mi enfermedad, algo tan desconocido y extraño, pero que forma parte de mi vida, de mí mismo.
Clasificación de las Enfermedades Raras
Hay que puntualizar que es muy difícil hacer esta clasificación, cuando hay tantas patologías, y con el problema añadido de que la mayoría son multisistémicas. Se pueden hacer clasificaciones atendiendo a distintos criterio: clasificaciones por herencia, por sistema o aparato fisiológico afectado, por causalidad de la patología, por prevalencia, etc. Atendiendo a la frecuencia de pacientes en la población podemos distinguir:
1. Enfermedades raras, más frecuentes aquellas con prevalencia entre de uno a cinco pacientes cada 10.000 habitantes: Neurofibromatosis, HHT.
2. Enfermedades raras no tan frecuentes, aquellas con una prevalencia entre uno y 20.000, y entre uno y 100.000: VHL.
3. Enfermedades ultrarraras, aquellas con prevalencia menor de uno cada 100.000 habitantes: Wolfram.
Agrupación de las Enfermedades Raras: grupos recomendados por EURORDIS
Se clasifican en 21 grupos atendiendo al órgano o aparato fundamentalmente, afectado:
1. Enfermedades de los huesos (Osteogénesis imperfecta).
2. Cánceres raros (Von Hippel Lindau, Paragangliomas, Feocromocitomas).
3. Enfermedades cardíacas: Síndrome de Holt-Oram.
4. Enfermedades de tejido conectivo y muscular (Sindrome de Ehler-Danlos).
5. Anomalías craneofaciales y de oído, nariz y garganta (Otorrinolaringología).
6. Enfermedades Endocrinas (Acondrodisplasia), Diabetes raras (Wolfram).
7. Retinopatías y otras afectaciones oculares raras: retinopatía de Leber, Amaurosis, Wolfram.
8. Enfermedades Gastrointestinales: síndrome de Hirschschprung).
9. Enfermedades ginecológicas y obstétricas : endometriosis,
10. Enfermedades hematológicas (Hemofilia, coagulopatías, sindrome urémico hemolítico).
11. Enfermedades hepáticas: Enfermedad de Caroli (dilatación quística congénita del árbol biliar intrahepática).
12. Enfermedades metabólicas: Gaucher, Leucodistrofía.
13. Enfermedades autoinmunes y del sistema inmunitario: Behçet, Sjögren, mastocitosis.
14. Malformaciones, anomalías del desarrollo y discapacidad intelectual: Angelman, autismos.
15. Enfermedades vasculares multisistémicas: Rendu-Osler, o HHT, síndrome SturgeWeber.
16. Enfermedades neurológicas: Epilepsias raras, Dravet, Wolfram, Narcolepsia.
17. Enfermedades Neuromusculares: Distrofias musculares, ELA.
18. Enfermedades Pulmonares: Fibrosis quística.
19. Enfermedades Renales: Síndrome urémico hemolítico.
20. Enfermedades de la piel (piel de mariposa, ictiosis, Epidermolisis bullosa).
21. Enfermedades del tracto genitourinario (Extrofia vesical).
Organización de las familias de afectados en asociaciones. FEDER
La mejor ayuda ante problemas grandes es asociarse, es un fenómeno adaptativo, propio de la sociedad humana.
FEDER (la Federación Española de Enfermedades Raras) nació en 1999 con seis asociaciones y hoy en día FEDER agrupa a más de 350 entidades (asociaciones, fundaciones y federaciones) siendo referente asociativo a nivel nacional de las enfermedades raras. Es el fenómeno social del empoderamiento de los pacientes ante su enfermedad, un problema que la sociedad y la Medicina normal no contempla, o no conoce lo suficiente.
FEDER representa a todas las enfermedades raras codificadas o sin codificar (código médico IE) y a las personas que están en espera de diagnóstico. Y FEDER apoya al diagnóstico de las enfermedades raras, a través de su Fundación FEDER.

¿Por qué investigar las Enfermedades Raras?
Porque es la única esperanza que les queda a las enfermedades raras.
Sólo un 10% de las ERs tienen algún tipo de investigación activa. De ellas tan sólo un 4% tienen algún tratamiento específico. La Federación Española de Enfermedades Raras (FEDER), a través de su Fundación, tiene como principal objetivo impulsar la investigación de las patologías minoritarias.
Sin investigación no hay futuro y por esta razón se necesita apoyar y fortalecer la investigación de nuestras entidades asociadas a través del impulso y desarrollo de becas.
¿Cómo se consigue el dinero?
Difusión en radio, prensa, televisión. Campañas SMS solidario. Crowdfunding, mecenazgo científico a base de pequeñas contribuciones solidarias on line. Conciertos solidarios.
Lotería, comercio solidario. Donativos públicos (empresas, bancos, responsabilidad social) y donaciones privadas.
Nuestra investigación en HHT
Desde el año 2002, emprendimos la tarea de empezar desde cero la investigación en el síndrome de Síndrome de Rendu-Osler-Weber (en honor a sus descubridores), también llamado Telangiectasia Hemorrágica Hereditaria, por sus síntomas clínicos, (HHT). Sabíamos que tenía que haber personas afectadas, pero no había nadie en España, ni médicos, ni investigadores, ni asociación de pacientes. Todo lo empezamos partiendo de “cero” (from the scratch, como dirían los americanos).
La HHT, es una enfermedad vascular, que se hereda con carácter dominante. Es decir, los pacientes son heterocigotos, tienen un gen normal y el otro mutado. Además, en una pareja en la que uno de los dos padres está afectado, un 50% de la descendencia tiene probabilidad de heredarla. Los genes que aparecen mutados en el 90% de los casos son dos receptores de la membrana de la célula endotelial: Endoglina (cromosoma 9) y la quinasa (ALK1) (cromosoma 12). Ambos forman el complejo receptor específico de la señalización de TGF-beta (factor de crecimiento transformante) en la célula endotelial, que tapiza la capa más interna de la vasculatura.
Es una enfermedad vascular autosómica dominante. La prevalencia estimada a nivel mundial es de un afectado cada 5.000 – 8.000, habitantes, pero más cerca de 5.000 que de 8.000. Se diagnostica clínicamente por los llamados criterios de Curaçao: epistaxis (sangrados nasales, espontáneos y recurrentes, que aumentan con la edad), telangiectasias (manchitas mucocutáneas en piel y mucosas), malformaciones arteriovenosas en órganos internos que se deben a una comunicación directa arteria-vena (en pulmón, hígado, cerebro, médula) y herencia dominante. Cuando una persona presenta tres de los cuatro criterios, podemos afirmar que es un paciente de HHT. Los criterios de epistaxis y telangiectasias, son evidentes desde los 25-30 años. La herencia familiar también. Lo que no resulta evidente, si no se hace una revisión médica exhaustiva que incluya análisis de imagen por RMN o TAC es la presencia de las malformaciones vasculares internas. Por eso es tan importante el hacer un diagnóstico genético precoz, para pasar a la revisión clínica y descartar la presencia de estas malformaciones. Y si las hubiera, “arreglarlas” por embolización (oclusión del punto de conexión al que se accede por un catéter con un dispositivo oclusivo en la punta).
Investigación traslacional en HHT
– Empezamos a finales del 2002 en el CIB, juntamente con dos médicos de medicina interna y un otorrino del hospital de Sierralla, hospital comarcal de Torrelavega, conectado con el Hospital Universitario Marqués de Valdecilla, Santander.
– En el 2003, se declara a Sierrallana hospital de referencia español para HHT por parte de la Fundación Internacional de HHT.
En el 2005 se registra formalmente la asociación HHT de pacientes y aparece la unidad de HHT España.
Ya teníamos las “tres patas” para sostener la “banqueta” de esta unidad: Pacientes, Médicos e Investigación. Han sido desde el 2002, hasta la actualidad 2016, casi 14 años, de trabajo fructífero, en el que se ha hecho diagnóstico genético, se han buscado las bases moleculares de la enfermedad y en los últimos años, nos hemos dedicado a la búsqueda de fármacos, que existiendo ya en la industria, concebidos para otros usos, pudieran ser efectivos en el tratamiento de los sangrados, nasal y digestivo de HHT.
Fruto de este trabajo, han sido no sólo las tesis y publicaciones, sino lo que es más importante tener dos medicamentos, que se usaban para la osteoporosis, considerados por la EMA (Agencia Europea del Medicamento), como medicamentos huérfanos para paliar las hemorragias derivadas de HHT.
Trabajamos por y para los pacientes, con una enorme vocación, empezando por los médicos y terminando por los investigadores. Ellos son nuestros mejores logros y resultados.
Imagen de la unidad de HHT España
Imagen de la unidad de HHT, que se asienta sobre los pilares de la asociación (los pacientes, HHT-España), el hospital de Sierrallana/Valdecilla (clínicos) y la investigación (CIB., CSIC).
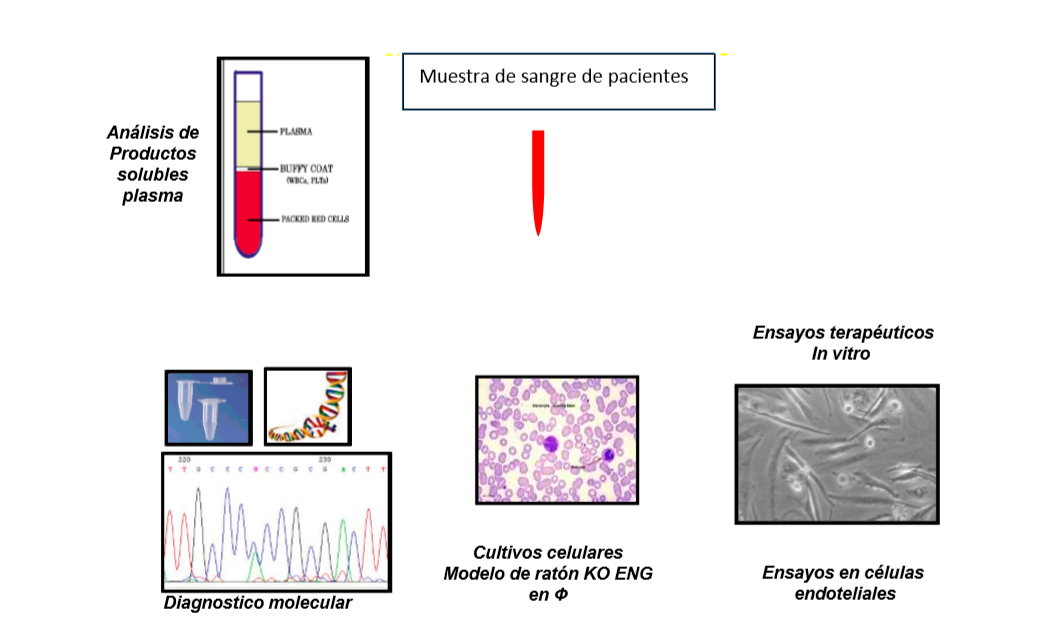
Estrategias terapéuticas para disminuir sangrados en HHT
Seguimos tres tipos de vías de experimentación para encontrar medicamentos que puedan controlar las epistaxis o los sangrados gastrointestinales:
1. Reforzar el proceso de coagulación, intentando disminuir las hemorragias por estabilización del coágulo de fibrina con antifibrinolíticos: Acido Tranexamico (Amchafibrín en España).
2. Estimular la expresión de los genes mutados en heterozigosis, para compensar la haploinsuficiencia de ENG y ALK1 en HHT. (SERMs moduladores específicos de los receptores estrogénicos: Raloxifeno (Evista), Bazedoxifeno (Conbriza)).
3. Antiangiogénesis: disminución de la vasculatura anormal y excesiva de las mucosas que provoca el sangrado hemorrágico (propranolol, timolol, en investigación el dobesilato/etamsilato, etc).
La familia crece: un nuevo miembro a investigar, la enfermedad de von Hippel Lindau
Justo hacia finales del 2012 y coincidiendo con el inicio de las terapias antiangiogénicas, la alianza von Hippel Lindau (VHL), nos contactó para estudiar la posible aplicación del propranolol como uso para frenar el desarrollo de tumores propios de esta enfermedad, y retrasar así las cirugías. La enfermedad de von Hippel Lindau, es una enfermedad rara (prevalencia de uno cada 36.000 habitantes), autosómica dominante, producida por la mutación en el gen VHL. Este gen es el encargado de controlar y degradar al factor de hipoxia en condiciones de crecimiento con oxigenación suficiente. El déficit en oxígeno, por debajo del 21%, causa el disparo del programa génico dirigido por el factor de hipoxia (HIF), programa que dispara también el proceso de desarrollo tumoral.
A partir del 2013 con apoyo de la alianza VHL, se incorporó el estudio de angiangiogénesis en cultivos primarios de hemangioblastomas y carcinomas derivados de las cirugías de estos pacientes. Ya se han obtenido resultados esperanzadores con el uso del propranolol y la posible repercusión en la detención del crecimiento de tumores cuyo factor desencadenante se el HIF.
Hipótesis de actuación del propranolol en las células
En resumen y para finalizar diríamos que nos encontramos con los problemas propios de la investigación de las enfermedades raras en España. Pero tenemos la vocación propia de la gente: médicos, asociaciones, investigadores que trabajan por y para los pacientes de estas enfermedades minoritarias. El esquema final, sea manifiesto visible de la situación real.
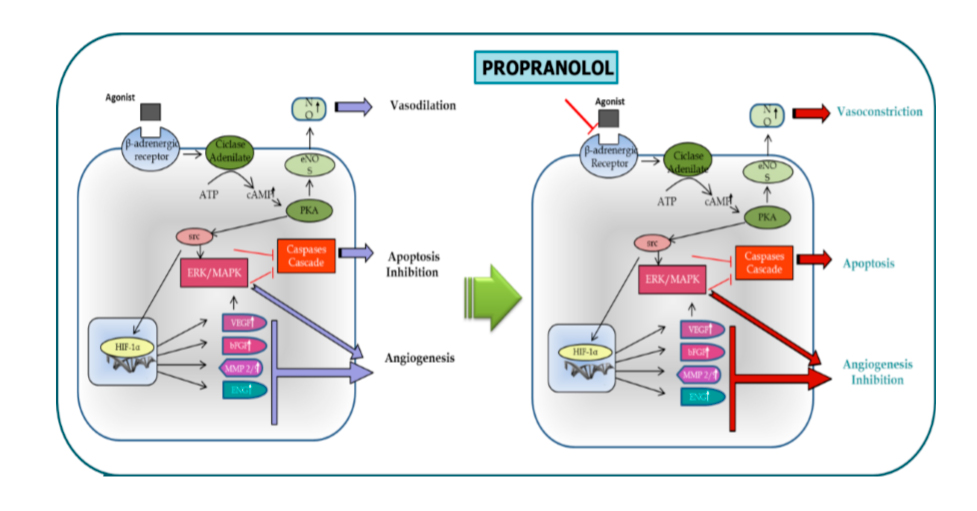
Hipótesis de actuación del propranolol en las células
En resumen y para finalizar diríamos que nos encontramos con los problemas propios de la investigación de las enfermedades raras en España. Pero tenemos la vocación propia de la gente: médicos, asociaciones, investigadores que trabajan por y para los pacientes de estas enfermedades minoritarias. El esquema final, sea manifiesto visible de la situación real.